
- About
- Organization
- Organization Overview
- Dean’s Office
- Department of Bioengineering and Therapeutic Sciences
- Department of Clinical Pharmacy
- Department of Pharmaceutical Chemistry
- Quantitative Biosciences Institute
- Org Chart
- Research
- Education
- Patient Care
- People
- News
- Events
Child’s illness fuels lab team’s search for early-life epilepsy diagnostics
By David Jacobson / Mon Jul 23, 2012

Postdoctoral fellow Ramon Birnbaum, PhD, and daughter Ruth.
The lab of UCSF School of Pharmacy faculty member Nadav Ahituv, PhD, studies how abnormalities in DNA segments that control the activity of genes could lead to diseases. Recently, researchers there have begun applying that focus to seeking a genetic basis and diagnosis for an epilepsy of early childhood, a disease that hit home for one lab scientist.
When Ramon Birnbaum, PhD, came to UCSF three years ago to do his postdoctoral work on the role of genetic regulation in human disease in the lab of School of Pharmacy faculty member Nadav Ahituv, PhD, epilepsy was barely on his radar.
Once, while serving in the Israeli military, he saw a fellow soldier suffer a seizure. Upon arriving on campus, just out of curiosity, he attended a presentation by Daniel Lowenstein, MD, director of the UCSF Epilepsy Center. That was the sum total of Birnbaum’s experience with this spectrum of seizure disorders, which affect an estimated 50 million people worldwide.
Then, in 2010, his daughter Ruth was born, and everything changed.
The parents of three healthy boys, Birnbaum and his wife, Adva, immediately noticed when three-week-old Ruth exhibited “subtle but unusual repetitive movements.” Following her intuition, Adva took their newborn to the pediatrician, who suspected Ruth was having seizures.
According to Birnbaum, the first neurologist they consulted thought it was probable that Ruth had Ohtahara syndrome, a severe disorder with a grim prognosis. Indeed, despite prescribed medications, Ruth’s seizure rate worsened, he recalls. She suffered several clusters per day, with some involving hundreds of convulsions over the course of an hour.
“It’s hard to imagine and even harder when, as a parent, you can’t do anything to help her,” Birnbaum says. “Her brain was on fire 24/7 with no chance to develop normally.”
But a second opinion by Joseph Sullivan, MD, director of the UCSF Pediatric Epilepsy Center, was more optimistic.
While abnormal electrical activity in Ruth’s brain was similar to that seen in Ohtahara, after viewing a homemade video of her seizures, Sullivan diagnosed her as having infantile spasms, a different type of early onset epilepsy. (See sidebar: Epilepsy and infantile spasms.)
Reviewing magnetic resonance imaging (MRI) of her brain, he also noted some abnormally located clumps of neurons within the normal overall brain structure (called gray matter heterotopia).
This diagnosis was key to selecting an effective therapy, since there are drugs that can treat infantile spasms, although they require careful dosing given a risk of significant side effects.
In Ruth’s case, her seizures completely disappeared within two months of starting and adjusting the drug therapy. She just turned two years old and, though experiencing some developmental delays, is “a laughing, smiling, crawling, interacting, and lovely baby,” says Birnbaum. She has not needed medications for nearly a year and a half.
From diagnosis to discovery
More than 170 years after infantile spasms entered medical literature—in a letter by British physician William West to The Lancet that was both a classic of clinical description and a poignant plea for help, since the patient was West’s son and there were no effective treatments then—Birnbaum, dealing with his daughter’s disorder, saw an unmet need for better diagnostic tools.
“Even with EEG and MRI results, which are not accessible for every baby, it is not always straightforward to diagnose this type of epilepsy,” says Birnbaum. Ruth had all the standard prenatal genetic testing as well as other “advanced genetic tests” in the wake of her seizures, all of which came back normal.
“We were lucky that our pediatrician sent us immediately to neurologists and that Ruth’s condition was correctly diagnosed, changing her life,” he says.
For Birnbaum and his advisor, Ahituv, a geneticist in the UCSF Department of Bioengineering and Therapeutic Sciences, the experience fueled a drive to discover a genetic diagnosis for infantile spasms, and potentially for other epilepsies and complex diseases.
Fresh fuel for that effort arrived in March, when Ahituv and Birnbaum were awarded a four-year, $1.1 million grant from the National Institutes of Health (NIH) to study whether mutations in gene regulatory elements—DNA segments that regulate the activity of genes—are a cause of infantile spasms and other epilepsies.
Their grant is part of the EUREKA (Exceptional Unconventional Research Enabling Knowledge Acceleration) program in the epilepsies. The program seeks to “move epilepsy research forward in leaps rather than in incremental steps” by supporting scientists “who want to test novel, unconventional hypotheses or pursue major methodological or technical challenges.”
A genetic diagnosis of infantile spasms and other epilepsies would allow for better tailored and faster application of current treatments for children like Ruth. The sooner that childhood epilepsies such as infantile spasms can be treated the better, notes Sullivan. Speed is crucial because as disruptive as daily seizures are, they reflect abnormal electrical activity in the brain that is going on round-the-clock and interfering with normal development.
“In those kids with no clear cause, if spasms stop and their EEG normalizes [with treatment], 50 to 60 percent can have normal outcomes,” says Sullivan.
Identifying variations in gene regulatory elements responsible for infantile spasms would increase understanding of the underlying mechanisms of the disease (see sidebar: Regulating gene expression). It could also provide the basis for new therapies that are more effective and precisely targeted, with fewer side effects.
Synergy with major UCSF epilepsy genome projects
Ahituv’s lab is positioned to make a unique contribution for several reasons:
- The lab not only researches gene regulatory elements but has a particular focus on examining how mutations in them could lead to human disease—including during development processes. For example, mutations in gene enhancers are a cause of limb malformations, the second most common human birth defect.
- Given the early onset of infantile spasms and the subset of patients in which there is no clear external cause or major brain/genetic abnormalities, the disorder poses a prime case for testing the role of mutations in gene regulatory elements involved in brain development.
- The Ahituv lab’s EUREKA project complements and benefits from two other major NIH-funded epilepsy studies currently being co-led by researchers at UCSF’s Epilepsy Center and UCSF School of Medicine’s Brain Development Research Program: the Epilepsy Phenome/Genome Project and Epi4K.
Since 2006, the on-going Epilepsy Phenome/Genome Project (EP/GP) has collected the DNA of nearly 4,000 patients along with details on their epilepsy (e.g., seizure types, EEG/MRI reports, therapy responses), including infantile spasms.
Starting last year, in a project dubbed Epi4K, 14 institutions worldwide are sequencing the EP/GP-collected genomes and analyzing them to look for associations between gene mutations (genotypes) and variations of the disease (phenotypes) as well as treatment response (pharmacogenomics).
Ultimately, Ahituv’s lab will receive genome sequence data from patients in those studies with infantile spasms and related conditions—as well as similar data on infantile spasm patients from their EUREKA collaborators at the Greenwood Genetic Center in South Carolina.
Instead of focusing on protein-encoding genes (as will be done in Epi4K), they will use these data to determine if there are mutations in those children’s genetic regulatory elements, specifically enhancers that are critically involved in normal brain development.
Seeking key regulators of brain development—and epilepsy
Indeed, the Ahituv lab’s research into gene regulation and infantile spasms will start by building on the current, still-limited knowledge of genetic factors that contribute to epilepsy.
“We have a list of about 100 genes that people have connected to epilepsy in some way,” says Ahituv.
These are not necessarily genes that have been shown to cause epilepsy in humans, he notes: “Some of these can be from mouse models [of the disease] … Others could have deletions or other chromosomal aberrations that do not include the gene itself but are near the gene.”
“Since we don’t know much about the genetic causes of epilepsy and infantile spasms, any epilepsy-associated gene could be a candidate,” says Ahituv. “And the first step of what we’re trying to do is to find out what regulates them in the brain during development … We’re trying to find specific regulatory elements for those genes.”
Initially, the scientists will analyze DNA from the forebrain tissue of a mouse embryo, which is genetically equivalent to the part of the developing human brain that is the source of seizures in infantile spasms.

Ahituv Lab
Overview of an enhancer assay
They will search this genome for tell-tale indications of enhancer activity— chemical modifications and proteins known as enhancer marks. They will use antibodies that bind to those marks allowing the enhancer-modified DNA segments to be culled, then sequenced and identified.
To confirm that the identified gene regulatory elements actually perform their roles in developing organisms, Ahituv and Birnbaum will inject those enhancers linked to a standard promoter and what is dubbed a reporter gene (see diagram) into fertilized zebrafish and mouse eggs.
If the enhancer activates an epilepsy-associated gene as the embryo develops, it will also activate the reporter gene whose expressed protein will be identifiable—glowing green under ultraviolet light or appearing blue when a stain is applied.
Finding and testing gene regulatory mutations in patients
In another year or two, the Ahituv lab will have access to the genomes from patients with infantile spasms collected by the two large UCSF co-led studies and the Greenwood Genetic Center. (Possibly including Ruth Birnbaum’s genome, although patient identifiers are removed from the genetic material to ensure individual privacy.)
Then, in their EUREKA project’s second phase, the scientists will look at those patients’ genomes for mutations in the human equivalents of the enhancers of epilepsy-associated genes that they found in the animal models. They will determine if the enhancers have mutations (nucleotide sequence variations) or are affected by copy number variations (deletions or multiplications of sections of DNA).
To confirm an active role for the enhancer variations found in patients with infantile spasms, Ahituv and Birnbaum will insert them into the fertilized eggs of animal models. They will measure whether the mutations change enhancer function, such as how well the enhancers bind to their activating transcription factors.
Compared to animals with normal genomes, the scientists will be able to see whether the mutant enhancers selected from infantile spasms patients are activating genes at the right location and at the correct time in an embryo’s brain development. Deviations in these key factors would indicate that mutations in these regulatory elements are involved in errors in brain development that may lead to infantile spasms.
Pharmacogenomics: The study of genetic variations in both patients and disease types that determine the effectiveness of a drug treatment is known as pharmacogenomics. It is a major research area of the Department of Bioengineering and Therapeutic Sciences, a joint department of the UCSF Schools of Pharmacy and Medicine and Ahituv and Birnbaum's home base.
Towards genome-based diagnosis and treatment
Ahituv and Birnbaum’s goal is to develop an overall picture of how gene regulatory elements work—or fail to work normally—in the developing brain in order to develop a genome-based diagnosis of infantile spasms and, potentially, other forms of epilepsy.
With the cost and time to perform complete genome sequencing plummeting, they foresee a day when testing for mutations in gene regulatory elements joins other standard genetic tests. Then, if an infant like Ruth Birnbaum develops a seizure disorder, physicians would know what it is sooner in order to treat it faster.
The complexity of epilepsy with its range of seizure types, onsets, severity, and treatment responses suggests that its causes, even for a developmental form like infantile spasms, may not be simple. However, this same clinical variation makes this research a model for understanding the role of gene regulation in other complex human diseases, such as cancer, autism, and heart disease.
“It could be not just one gene, but a few gene mutations that lead to epilepsies,” says Ahituv. “It could be not just one gene, but a few [abnormal] regulatory elements…. it could be a few regulatory elements and genes together such that the sum of these mutations leads to these phenotypes.”
Eventually, large-scale pharmacogenomic studies could determine the best therapies based on a given child’s sequence variations. “I’m not saying that we’re going to have that tomorrow,” says Ahituv. “This is our vision.”
Tags
Keywords:
epilepsy, UCSF Epilepsy Center, UCSF Pediatric Epilepsy Center, infantile spasms, genetic diagnosis, National Institutes of Health (NIH), genetics, EUREKA (Exceptional Unconventional Research Enabling Knowledge Acceleration), genetic mutations, Congenital Limb Deformities, UCSF Brain Development Research Program, Epilepsy Phenome/Genome Project, Epi4K, pharmacogenomics, enhancers, promoter, silencers, insulators, reporter gene, genome-based diagnosis, EEG (electroencephalography), magnetic resonance imaging (MRI)
Category:
Sites:
School of Pharmacy, Department of Bioengineering and Therapeutic Sciences, PharmD Degree Program, QBC, PSPG, Bioinformatics, BMI
About the School: The UCSF School of Pharmacy aims to solve the most pressing health care problems and strives to ensure that each patient receives the safest, most effective treatments. Our discoveries seed the development of novel therapies, and our researchers consistently lead the nation in NIH funding. The School’s doctor of pharmacy (PharmD) degree program, with its unique emphasis on scientific thinking, prepares students to be critical thinkers and leaders in their field.
Epilepsy and infantile spasms
Epilepsy “encompasses a number of different syndromes whose cardinal feature is a predisposition to recurrent unprovoked seizures,” wrote Lowenstein in a review article he co-authored for The New England Journal of Medicine.
The syndromes are classified by “the type of seizure, the presence or absence of neurologic or developmental abnormalities, and electroencephalographic (EEG) [brain electrical activity] findings,” he notes.
Some epilepsies are caused by head injuries, infections, strokes, and brain tumors. But others appear to be related to genetic predispositions and/or developmental brain anomalies.
Infantile spasms are both a type of seizure associated with and often a term considered synonymous with West Syndrome, a type of epilepsy characterized by four factors:
- Early onset, typically starting in the first year of life
- Seizure type—clusters of body-bending convulsions, although they can also be very subtle
- Specific types of abnormal EEG patterns
- Developmental regression
Infantile spasms can be symptomatic, meaning it is linked to:
- Infections and injuries before, during, or shortly after birth.
- Major brain-structure or genetic abnormalities, such as cortical dysplasia or Down Syndrome.
- Heterotopia, which may or may not play a causal role.
But nearly a third of cases have no clear causes, suggesting roles for mutations in genes and/or gene regulatory elements.
Regulating gene expression
Gene regulatory elements are segments of DNA that instruct genes when, where, and to what extent to turn on or off (making mRNA, which makes proteins).
In fact, genes, which provide the blueprints that are copied to make protein molecules—make up less than two percent of our DNA. The rest is noncoding, which includes things such as regulatory elements.
Like genes, the function of any given gene regulatory element is determined by the specific sequences of nucleotides that comprise it.
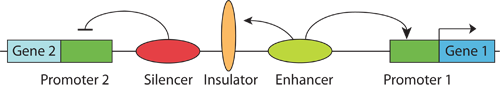
Gene regulatory elements
Julia VanderMeer
Gene regulatory elements are activated when proteins called transcription factors bind to them. Categorized by their effect on genes, they include:
- Promoters—serve as the site where enzymes and other proteins bind and assemble to start making RNA from DNA, the first step in gene expression (i.e., the creation of protein molecules).
- Enhancers—activate promoters at specific locations, times, and levels.
- Silencers—turn off gene expression at specific locations, and times.
- Insulators— prevent one gene’s regulatory elements from affecting its neighbors.
Enhancers are not necessarily near promoters and can be located anywhere in the genome. By contrast, promoters are adjacent to the genes they regulate. Once activated, enhancers interact with promoters, sometimes through a change in DNA shape called looping.
Activated enhancers will form a complex with promoters, transcription factors, and co-factors. The end result allows for the binding of proteins that then begin the process of transcribing a given gene’s blueprints to RNA, ultimately manufacturing a new protein.
Related news
- Ahituv Lab research finds how bats grow wings, with implications for human limb development
- Ahituv study provides new insights into enhancer gene regulation
- Ahituv appointed director of UCSF Institute for Human Genetics
- CRISPR-based obesity treatment shows promise for other diseases
- UCSF School of Pharmacy leads in NIH funding for 38th year


